Neuronal mechanisms of neuropathic pain after nerve injury. Mechanisms whereby injury evokes sensory dysfunction and chronic pain. Pathophysiology of injured nerve, particularly, abnormal discharge of electrical activity originates at the site of nerve injury or in the DRG in experimental animal models for neuropathy in the rat.
Relationship between sensory and motor modalities in the oro-facial area. Effects and mechanisms of different drugs and hypnosis on pain and reflexes in the oro-facial area.
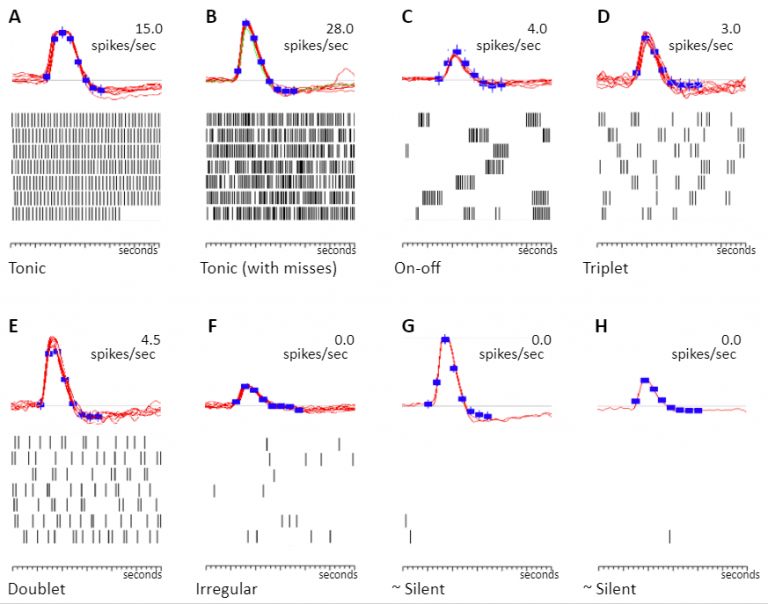
Resurgent neuropathic discharge: An obstacle to the therapeutic use of neuroma resection?
Ectopic discharge (“ectopia”) in damaged afferent axons is a major contributor to chronic neuropathic pain. Clinical opinion discourages surgical resection of nerves proximal to the original injury site for fear of resurgence of ectopia and pain with an intensity even greater than before. We tested this concept in a well-established animal neuroma model.
Genotype-selective phenotypic switch in primary afferent neurons contributes to neuropathic pain.
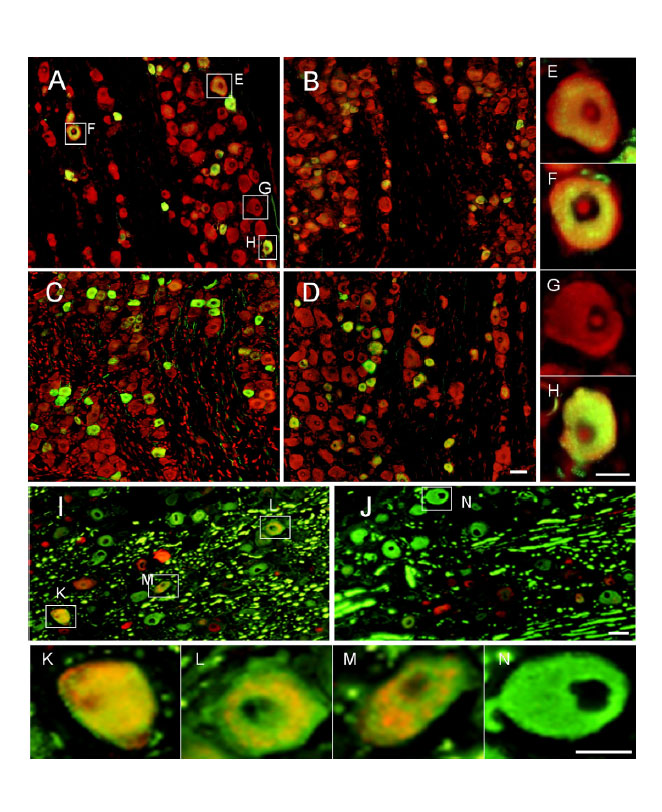
Pain is normally mediated by nociceptive Aδ- and C-fibers while Aβ-fibers signal touch.However, after nerve injury Aβ-fibers may signal pain. Using a genetic model we tested the hypothesis that phenotypic switching in neurotransmitters expressed by Aβ afferents might account for heritable differences in neuropathic pain behavior.
(Genotype-selective phenotypic switching in the dorsal root ganglion (DRG) (calcitonin gene-related peptide [CGRP]). Image: Adi Nitzan-Luques)
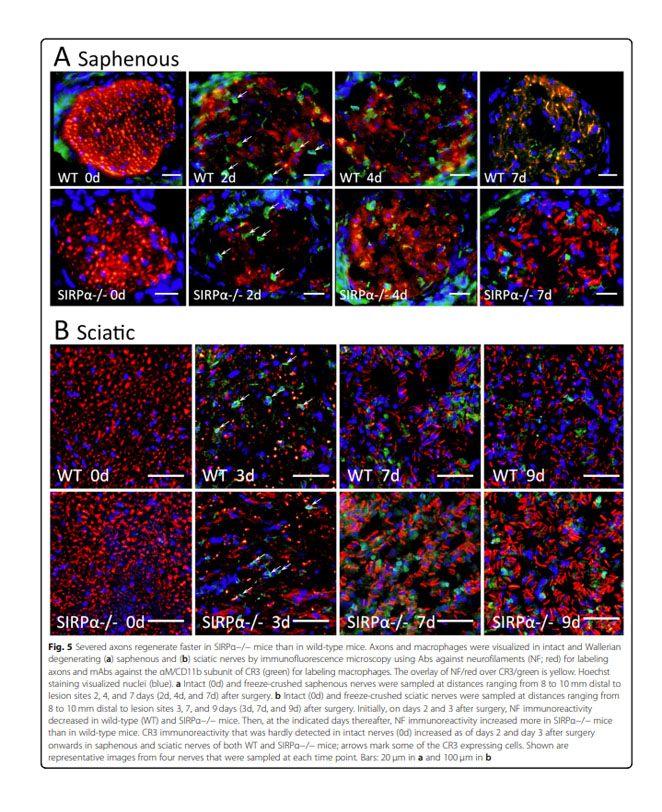
Deletion of SIRPα (signal regulatory protein-α) promotes phagocytic clearance of myelin debris in Wallerian degeneration, axon regeneration, and recovery from nerve injury.
Recovery of function from traumatic nerve injury depends on the ability of severed axons to grow/regenerate back to their target tissues. This is achieved by successfully crossing the lesion site where physical impact severed axons, determined by the type of trauma, followed by successfully growing throughout the Wallerian degenerating nerve segment located distal to and beyond the lesion site, determined by the nature of Wallerian degeneration. The protracted removal of myelin debris in Wallerian degeneration, which leads residual myelin debris to slow down axon growth, impedes recovery of function. We focused in this study on mechanism(s) that delay the removal of myelin debris in Wallerian degeneration and so impede recovery. We aimed to determine whether SIRPα-dependent inhibition of phagocytosis in macrophages impedes the in vivo removal of myelin debris in Wallerian degeneration, further leading to impaired healing.
Adi Nitzan-Luques, Marshall Devor and Michael Tal. (2011) Genotype-selective phenotypic switch in primary afferent neurons contributes to neuropathic pain. Pain 152: 2413-2426.
Our findings suggest that phenotypic switching contributes to the heritable difference in pain behavior in high-autotomy (HA) vs low-autotomy (LA) rats. Specifically, we propose that in HA rats, but less so in LA rats, injured, spontaneously active Aβ afferents both directly drive CGRP-sensitive central nervous system pain-signaling neurons and also trigger and maintain central sensitization, hence generating spontaneous pain and tactile allodynia.
Adi Nitzan-Luques, Marshall Devor and Michael Tal. (2013) Dynamic genotype-selective “phenotypic switching” of CGRP expression contributes to differential neuropathic pain phenotype. Exp. Neurol. 250:194-204.
First, we show that the time of emergence of CGRP-IR in DRG neurons and their central terminals matches that of pain behavior. Then we show, using FOS-IR as a marker, that following SNL injury activity in Aβ afferents is indeed capable of driving postsynaptic pain-signaling neurons and inducing central sensitization, and that the effect is much more prominent in HA than in LA rats. Finally, we show that a CGRP-receptor (CGRP-R) antagonist administered intrathecally (i.t.) attenuates SNL-evoked tactile allodynia although it does not block baseline nociception. Each of these lines of evidence supports the conclusion that genotype-selective phenotypic switching of CGRP expression in Aβ afferents following nerve injury is a fundamental mechanism of neuropathic tactile allodynia.
Marshall Devor and Michael Tal. (2014) Nerve resection for the treatment of chronic neuropathic pain. Pain 155: 1053-1054.
Nadav Y. Ziv, Michael Tal, Yehuda Shavit. (2016) The transition from naïve to primed nociceptive state: A novel wind-up protocol in mice. Experimental Neurol. Jan;275 Pt 1:133-42.
The present study employed a PoW paradigm consisting of three successive trains of electrical stimulation in mice, while recording EMG responses of the withdrawal reflex. Using this paradigm we sought to answer several questions: 1) What are the parameters required to produce the PoW response? 2) How does exposure to a PoW paradigm affect pain behavior? 3) Does the PoW response differ in naïve animals (without any prior noxious input) vs. noxious-primed animals; i.e., does prior exposure to “natural” noxious stimuli, such as heat or deep muscle trauma, affect the PoW response? And 4) The role of NMDA-receptors in this phenomenon.
Tal Michael , Villanueva L., Devor M. (2015) Anatomy and Neurophysiology of Orofacial Pain
in Orofacial Pain and Headache edts. Y. Sharav and R. Benoliel. Quintanses International.
Gerard Elberg; Sigal Liraz-Zaltsman; Fanny Reichert; Takashi Matozaki; Michael Tal; Shlomo Rotshenker. (2020) Deletion of SIRPα (signal regulatory protein-α) promotes phagocytic clearance of myelin-debris in Wallerian degeneration, axon regeneration, and recovery from nerve injury. Journal of Neuroinflammation. 16:277-290.
Our findings suggest an intrinsic normally occurring SIRPα-dependent mechanism that impedes the
in vivo removal of myelin debris in Wallerian degeneration by inhibiting the phagocytosis of myelin debris in macrophages, hence preventing fast growing axons from fully implementing their regenerative potential. Thus, accelerating the removal of myelin debris by eliminating SIRPα-dependent inhibition of phagocytosis will most likely advance recovery of functions from nerve injury.
Michael Tal
Prof. Tal is a laboratory head and the past President of The Israel Pain Association. Head of the Center for Research on Pain. Chair of The University Committee for the Use of Human Subjects in Research (IRB). Higher education was at Hebrew University (DMD) and UCLA (MS). He had worked for 2 years with Gary Bennett at NIH and later with Ron Dubner and Ken Ren In The University of Maryland Baltimore. Spent a sabbatical in the Department of Neurosurgery Johns Hopkins, Baltimore MD, working on the mechanisms of neuropathic pain. Visiting Scholar, NIH Department of Bioethics. His laboratory has published in the pain field of neuropathic pain, with work of a notably integrative nature involving neurophysiology, psychophysiology and animal behavior under chronic pain conditions.